Research
The Danko lab studies how our DNA sequences control complex programs of gene transcription. Our work is primarily focused on understanding how natural genetic differences between species affect the various steps in the RNA polymerase II transcription cycle. Our work provides insight into the molecular basis behind phenotypic differences between species. In addition, studying the millions of naturally occurring random genetic mutation “experiments” also presents an exciting opportunity to understand the fundamental principles by which our DNA sequences encode gene expression.
We are an interdisciplinary research group making extensive use of both computational and molecular tools. Our specialty is developing statistical and machine-learning approaches to analyze functional genomic sequencing data prepared using Hi-C, ATAC-seq, PRO-seq, RNA-seq, and related assays. Our tools borrow a variety of ideas from the fields of statistics and machine-learning, including recent uses of hidden Markov models, support vector machines, and artificial neural networks. We also run an active wet-lab that has made great strides in developing and using run-on and sequencing technologies to map the location of RNA polymerase, including PRO-seq and ChRO-seq. More recently we have begun using Hi-C/ Hi-ChIP, single cell RNA-seq, and CRISPR epigenome editing technologies.
Understanding the chain of molecular events that link DNA sequence ‘genotype’ to organism ‘phenotype’ is one of the most exciting frontiers in modern genetics. DNA sequences located in non-coding regions of the genome are critical drivers of phenotypic differences, both between and within species.
Our objective is to discover the fundamental rules by which transcriptional changes arise from differences in DNA sequence and chromatin packaging within the nucleus. To achieve this goal we integrate genomic data collected using a combination of molecular assays (PRO-seq, RNA-seq, ATAC-seq, and Hi-C). Most of our work focuses on CD4+ T-cells, a lynchpin in the adaptive immune system undergoing rapid evolutionary changes that are relevant to autoimmune and allergic disorders.
We previously found that although changes in distal regulatory elements arise rapidly, these changes frequently do not lead to measurable differences in the transcription of nearby genes. We found evidence that gene transcription is stabilized by multiple compensatory changes acting across ensembles of distal enhancers. This finding suggests a model of regulatory evolution in which changes in regulatory activities arise rapidly, and gene expression is held constant through widespread compensation between regulatory elements targeting each gene.
Detecting biochemically active DNA sequences in a cell (one of the common definitions of a cell's “epigenome”) is a major challenge in genomics. Many approaches rely on using dozens of separate experimental assays, making the analysis of new cell systems expensive and time-consuming. We have recently demonstrated that RNA polymerase marks a surprisingly broad variety of functional elements across the genome. These functional elements can be recognized based on their characteristic “shapes” extracted from PRO-seq data using machine learning tools.
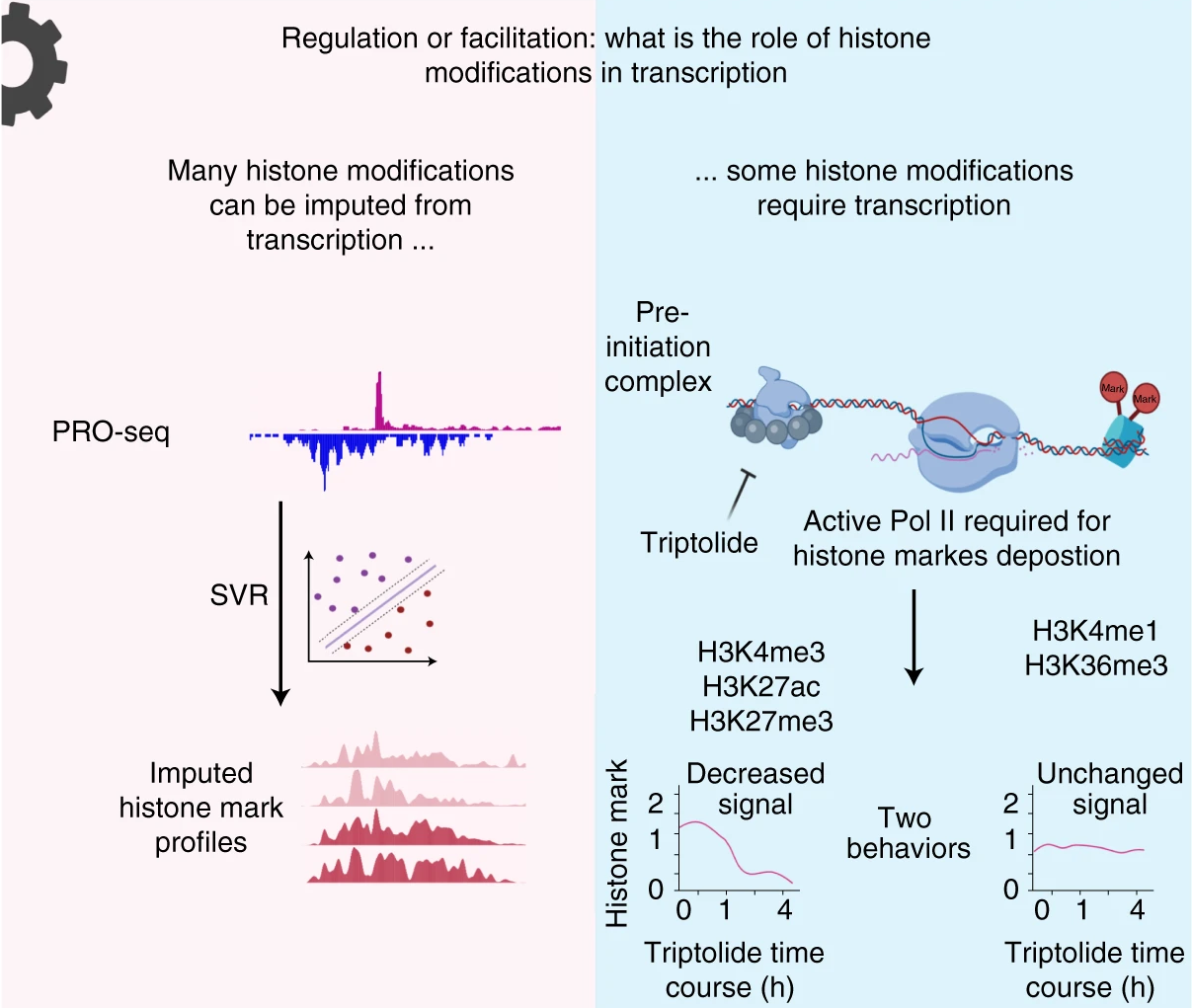
Our objective is to develop a computational toolkit that deconvolves a single PRO-seq assay into a rich source of information about multiple ‘layers’ of functional elements that are active in our genomes. We have developed a machine learning tool called dREG which identifies the location of active regulatory DNA sequence elements using PRO-seq data as input. More recently, we introduced dHIT, a discriminative support vector regression to guess or ‘impute’ the abundance of covalent modifications to core histones. These technologies allow comprehensive annotation of active functional elements in mammalian genomes using PRO-seq data alone.
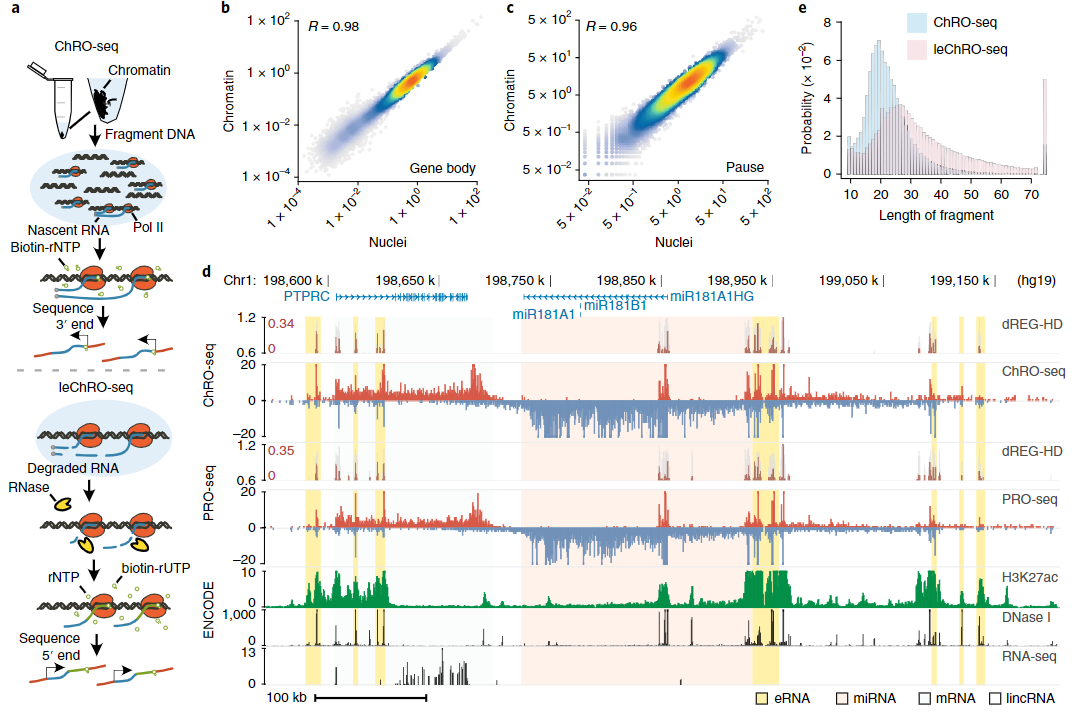
Finally, a core mission of the wet-lab is to extend run-on and sequencing assays to map the location of RNA polymerase across the genome in a wider range of biological conditions. We have recently introduced a new run-on and sequencing variant called ChRO-seq to address the key problem with PRO-seq: namely that it requires a nuclear isolation, which can be challenging in complex tissue samples such as muscle or brain. We have also made substantial progress on strategies to multiplex the PRO-seq and ChRO-seq assays using a 96-well plate format. Taken together, our efforts significantly expand the scope and range of applications in which PRO-seq can be applied.
RNA polymerase II dynamics shape enhancer-promoter interactions.
Barshad G, Lewis JJ, Chivu AG, Abuhashem A, Krietenstein N, Rice EJ, Ma Y, Wang Z, Rando OJ, Hadjantonakis AK, Danko CG.
Nature Genetics. (2023).
Genetic Dissection of the RNA Polymerase II Transcription Cycle.
Chou SP, Alexander AK, Rice EJ, Choate LA, Danko CG.
eLife. (2022).
Cell type and gene expression deconvolution with BayesPrism enables Bayesian integrative analysis across bulk and single-cell RNA sequencing in oncology.
Chu T, Wang Z, Pe'er D, Danko CG.
Nature Cancer. (2022).
Prediction of histone post-translational modification patterns based on nascent transcription data.
Wang Z, Chivu AG, Choate LA, Rice EJ, Miller DC, Chu T, Chou S, Kingsley NB, Petersen JL, Finno CJ, Bellone RR, Antczak DF, Lis JT, Danko CG.
Nature Genetics. (2022).
Multiple stages of evolutionary change in anthrax toxin receptor expression in humans.
Choate LA, Barshad G, McMahon PW, Said I, Rice EJ, Munn PR, Lewis JJ, Danko CG.
Nature Communications (2021).
Identification of regulatory elements from nascent transcription using dREG.
Wang Z, Chu T, Choate LA, Danko CG.
Genome Research (2019).
Chromatin run-on and sequencing maps the transcriptional regulatory landscape of glioblastoma multiforme.
Chu T, Rice EJ, Booth GT, Salamanca HH, Wang Z, Core LJ, Longo SL, Corona RJ, Chin LS, List JT, Kwak H, Danko CG.
Nature Genetics (2018).
Dynamic evolution of regulatory element ensembles in primate CD4+ T cells.
Danko CG, Choate LA, Marks BA, Rice EJ, Wang Z, Chu T, Martins AL, Dukler N, Coonrod SA, Tait-Wojno E, List JT, Kraus WL, Siepel A.
Nature Ecology & Evolution (2018).
A unified architecture of transcriptional regulatory elements.
Andersson R, Sandelin A, Danko CG.
Trends in Genetics (2015).
Identification of active transcriptional regulatory elements from GRO-seq data.
Danko CG, Hyland SL, Core LJ, Martins AL, Waters CT, Lee HW, Cheung VG, Kraus WL, Lis JT, and Siepel A.
Nature Methods (2015).